Tumor Cells' 'Ferroaddiction' Can Be Their Achilles Heel
Metals pervade all aspects of the central dogma because they are essential for the function of all DNA and RNA molecules, and one-third to one-half of all proteins, in addition to contributing to epigenetic modifications. The recent emergence of signaling roles for transition metals presages a broader contribution of these elements beyond their traditional functions as metabolic cofactors. Connections between transition-metal metabolism and signaling have emerged recently in the cancer field. An elegant example of transition-metal signaling in cancer has identified a new form of physiology via iron-dependent cell death, termed ferroptosis. This cell death pathway was uncovered in the course of studying the mechanism of action of erastin , a small molecule that selectively kills cells expressing oncogenic mutants of RAS. A previous study has found elevated Fe2+ is linked by an unknown mechanism to a drug-tolerant cancer cell state that is also highly susceptible to ferroptosis. Taken together, these studies suggest that access to bioavailable Fe2+ (as opposed to total iron) is an important factor in cancer cell survival and proliferation.
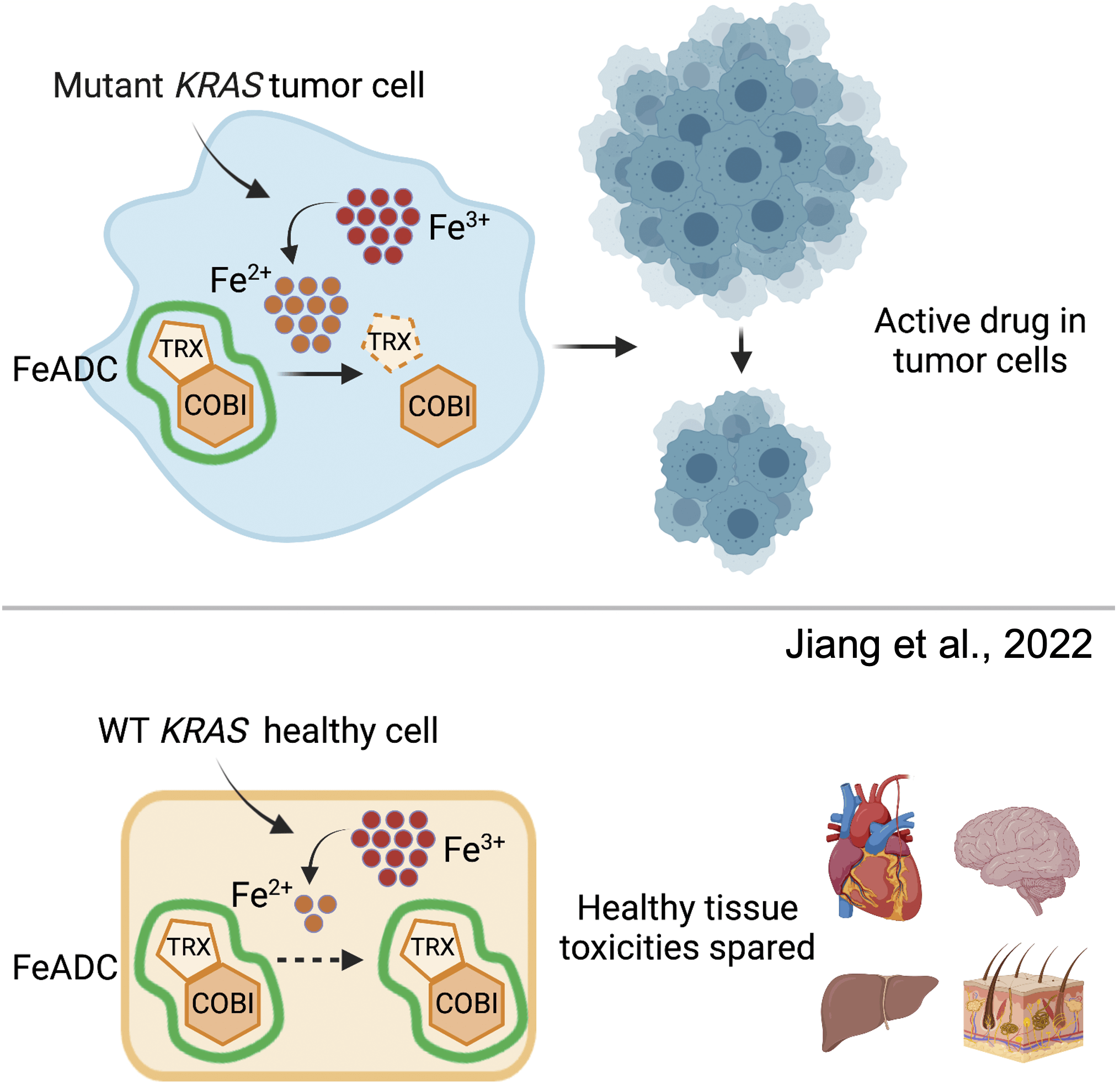
A new study from researchers at the University of California, San Francisco (UCSF) has found that oncogenic KRAS signaling induced ferrous iron (Fe2+) accumulation early in and throughout mutant KRAS-mediated transformation. Based on this, they converted an FDA-approved MEK inhibitor into a ferrous iron–activatable drug conjugate (FeADC) and achieved potent MAPK blockade in tumor cells while sparing normal tissues. This innovation allowed sustainable, effective treatment of tumor-bearing animals, with tumor-selective drug activation, producing superior systemic tolerability.
To evaluate tumor iron metabolism in an unbiased manner, the researchers first evaluated mRNA transcriptional data from 23 tumor types profiled by The Cancer Genome Atlas (TCGA) with a focus on iron homeostatic programs using biological pathway activity method. The cancer types with the highest Z-scores for iron metabolism were all RAS–MAPK pathway-driven malignancies, which include glioblastoma multiforme, lung adenocarcinoma, and pancreatic ductal adenocarcinoma (PDA). Focusing on PDA specifically, they found that this increased iron concentration was correlated with shorter survival times in patients. To assess iron avidity in PDA, they conducted an exploratory human imaging study with Ga-68 citrate positron emission tomography (PET) in our PDA patients. Quantitative analysis of the imaging data showed that visceral and bony PDA metastases were highly avid for Gallium-68 compared to normal tissues, which is consistent with an iron avid phenotype in PDA.
Evaluation of a larger panel of human malignant cell lines from PDA and breast cancer confirmed that cells harboring mutant KRAS had consistently higher levels of total intracellular ferrous iron. The UCSF team hypothesized that it might be possible to utilize this feature of PDA cancer cells to their advantage. Could they be selectively targeted by drugs that are activated by ferrous iron, termed ferrous iron-activatable drug conjugates (FeADCs)? FeADCs are inactive versions of drugs that break apart in the presence of Fe2+, releasing the drug's active version. The approach was inspired by anti-malarial drugs like artemisinin that target Fe2+ in the parasite as it invades red blood cells and degrades hemoglobin, producing large amounts of free heme iron.
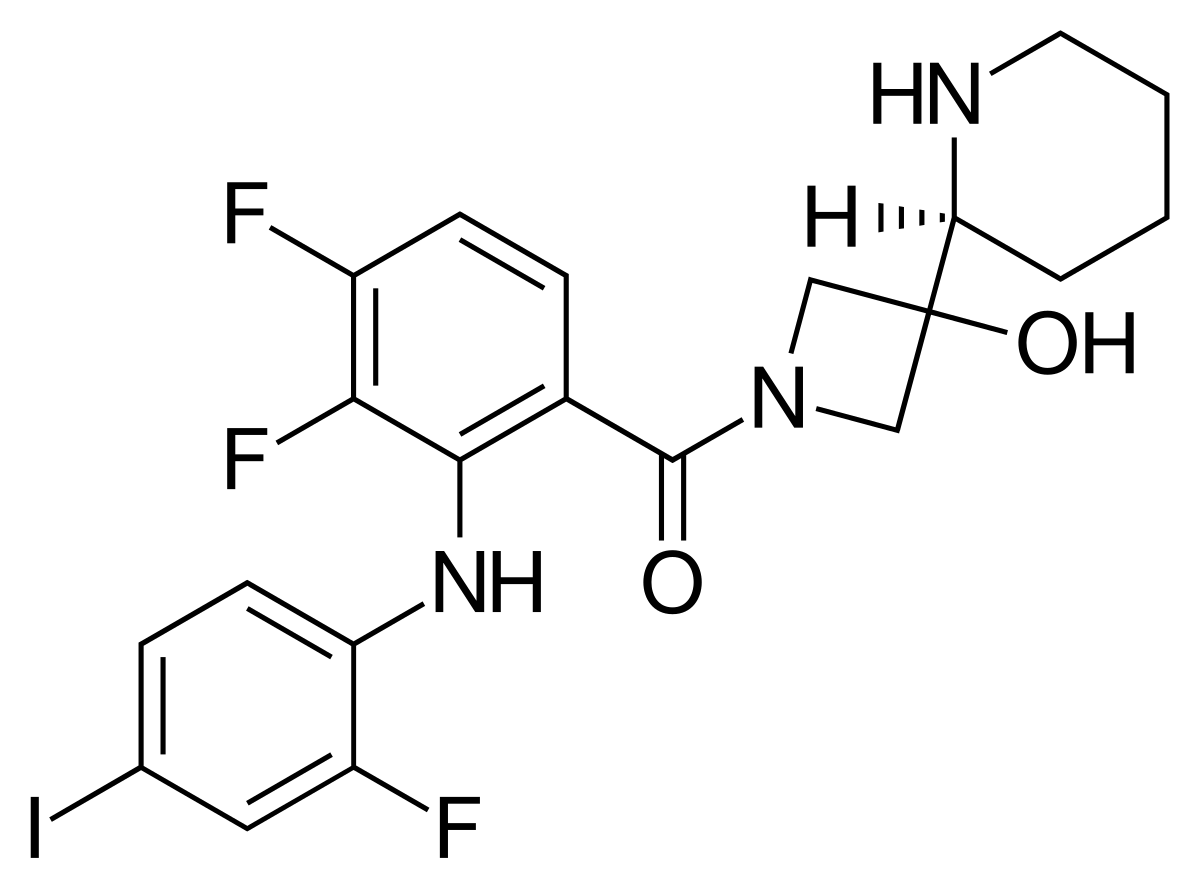
To determine whether a FeADC approach might be effective in mutant KRAS-driven malignancies, the researchers studied PDA cells with the Fe2+ probe TRX-PURO, a Fe2+-activatable conjugate of the aminonucleoside puromycin. Having established that mutant RAS signaling elevates intracellular Fe2+, as measured with both SiRhoNox and TRX-PURO, we next sought to target the RAF-MEK-ERK pathway in PDA with a conditionally activated (FeADC-type) inhibitor of this pathway. We hypothesized that a FeADC targeting MEK1/2 would be efficiently activated by Fe2+ in oncogenic KRAS-driven tumors, but not in healthy mouse epidermis, where normal iron homeostasis is preserved. We synthesized FeADCs bearing the FDA-approved allosteric MEK inhibitor cobimetinib (COBI) and based on either enantiomeric form of the Fe2+-targeting TRX moiety, yielding the novel agents (R,R)-TRX-COBI and (S,S)-TRX-COBI. To evaluate the FeADC approach to MEK1/2 inhibition in vivo, the researchers orthotopically implanted mouse KrasLSL_G12D, Trp53f/+, Pdx1Cre (KPC) cells into syngeneic FVB mice and treated with equimolar doses of either TRX-COBI or COBI. Both drug forms were comparably growth inhibitory and showed equivalent suppression of phospho-ERK levels, at equimolar doses in this orthotopic PDA model.
Through a series of in vitro and in vivo experiments, the researchers tested whether the FeADC would be able to disrupt KRAS signaling pathways and block cancer cell growth, without eliciting adverse effects to healthy cells and tissues. The in vivo approaches used mouse models of KRAS-driven cancer, like PDA and lung adenocarcinoma. Across the in vitro and in vivo experiments, the scientists found that TRX-cobimetinib inhibited tumor growth to the same standard as cobimetinib without causing damage to healthy cells. As such, they combined TRX-cobimetinib with other anticancer drugs and discovered that the combination of therapeutics was even more effective at inhibiting tumor growth, again, without adverse side effects. Across the in vitro and in vivo experiments, the scientists found that TRX-cobimetinib inhibited tumor growth to the same standard as cobimetinib without causing damage to healthy cells. As such, they combined TRX-cobimetinib with other anticancer drugs and discovered that the combination of therapeutics was even more effective at inhibiting tumor growth, again, without adverse side effects.
More about KRAS
KRAS (Kirsten rat sarcoma virus) is a gene that provides instructions for making a protein called K-Ras, a part of the RAS/MAPK pathway. It is called KRAS because it was first identified as a viral oncogene in the Kirsten RAt Sarcoma virus. The protein relays signals from outside the cell to the cell's nucleus. These signals instruct the cell to grow and divide (proliferate) or to mature and take on specialized functions (differentiate).
Mutations in KRAS are found in many cancers and are particularly common in pancreatic ductal adenocarcinoma (PDA), colorectal cancer, acute myeloid leukemia, and lung adenocarcinoma. In total, KRAS mutations are thought to cause a quarter of all cancer deaths by activating cell signaling pathways that drive cell proliferation and enhance cell survival. These signaling pathways can be blocked by drugs that inhibit some of the proteins activated by KRAS, but, in addition to killing cancer cells, these drugs are highly toxic to healthy cells and tissues, limiting their use at doses needed to inhibit signaling in cancer cells.
In July 2009, the US Food and Drug Administration (FDA) updated the labels of two anti-EGFR monoclonal antibody drugs indicated for treatment of metastatic colorectal cancer, panitumumab (Vectibix) and cetuximab (Erbitux), to include information about KRAS mutations.
In 2012, the FDA also cleared QIAGEN's therascreen KRAS test, which is a genetic test designed to detect the presence of seven mutations in the KRAS gene in colorectal cancer cells. This test is used to aid physicians in identifying patients with metastatic colorectal cancer for treatment with Erbitux. The presence of KRAS mutations in colorectal cancer tissue indicates that the patient may not benefit from treatment with Erbitux. If the test result indicates that the KRAS mutations are absent in the colorectal cancer cells, then the patient may be considered for treatment with Erbitux.
Other drugs being tested with KRAS as a drug target
More about MAPK pathway
The MAPK/ERK pathway (also known as the Ras-Raf-MEK-ERK pathway) is a chain of proteins in the cell that communicates a signal from a receptor on the surface of the cell to the DNA in the nucleus of the cell. The pathway includes many proteins, such as mitogen-activated protein kinases (MAPKs), originally called extracellular signal-regulated kinases (ERKs), which communicate by adding phosphate groups to a neighboring protein (phosphorylating it), thereby acting as an "on" or "off" switch. MAPK cascades are central signaling elements that regulate basic processes including cell proliferation, differentiation and stress responses.
Facts about pancreatic ductal adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC) is the most prevalent neoplastic disease of the pancreas accounting for more than 90% of all pancreatic malignancies. To date, PDAC is the fourth most frequent cause of cancer-related deaths worldwide with a 5-year overall survival of less than 8%. The incidence of PDAC is expected to rise further in the future, and projections indicate a more than two-fold increase in the number of cases within the next ten years, both in terms of new diagnoses as well as in terms of PDAC-related deaths in the U.S. as well as in European countries. Life style habits, including alcohol and tobacco abuse, which are well-known to increase the risk for several other types of cancer, such as lung cancer and squamous cell carcinomas of the head and neck region, also appear to be involved in PDAC development. Finally, for a subgroup of approximately 5-6% of all PDAC patients, genetic predispositions, such as germline mutations in the genes BRCA1/2, ATM, MLH1, TP53, or CDKN2A, represent further risk factors.
References:
- Chang, Christopher J. "Searching for harmony in transition-metal signaling." Nature chemical biology 11, no. 10 (2015): 744-747.
- Honglin Jiang, Ryan K. Muir, Ryan L. Gonciarz, Adam B. Olshen, Iwei Yeh, Byron C. Hann, Ning Zhao, Yung-hua Wang, Spencer C. Behr, James E. Korkola, Michael J. Evans, Eric A. Collisson, Adam R. Renslo; Ferrous iron–activatable drug conjugate achieves potent MAPK blockade in KRAS-driven tumors. J Exp Med 4 April 2022; 219 (4): e20210739.