Study Looks into RASRx1902’s Effects on Slowing Progression of ALS-Related Muscle Degeneration
Amyotrophic lateral sclerosis (ALS) was once commonly known as Lou Gehrig’s disease, following the retirement of the famous ballplayer in the 1940s due to the disease. ALS is a rare neurological disease that primarily affects the nerve cells (neurons) responsible for controlling voluntary muscle movement. Voluntary muscles produce movements like chewing, walking, and talking. The disease is progressive, meaning the symptoms get worse over time. Currently, there is no cure for ALS and no effective treatment to halt or reverse the progression of the disease. ALS is the most common type of motor neuron disease. Early symptoms of ALS include stiff muscles, muscle twitches, and gradual increasing weakness and muscle wasting. Motor neuron loss continues until the ability to eat, speak, move, and finally the ability to breathe is lost. ALS eventually causes paralysis and early death, usually from respiratory failure.
ALS occurs throughout the world with no racial, ethnic, or socioeconomic boundaries. It affects as many as 30,000 in the United States, with 5,000 new cases diagnosed each year. Estimates suggest that ALS is responsible for as many as five of every 100,000 deaths in people aged 20 or older.
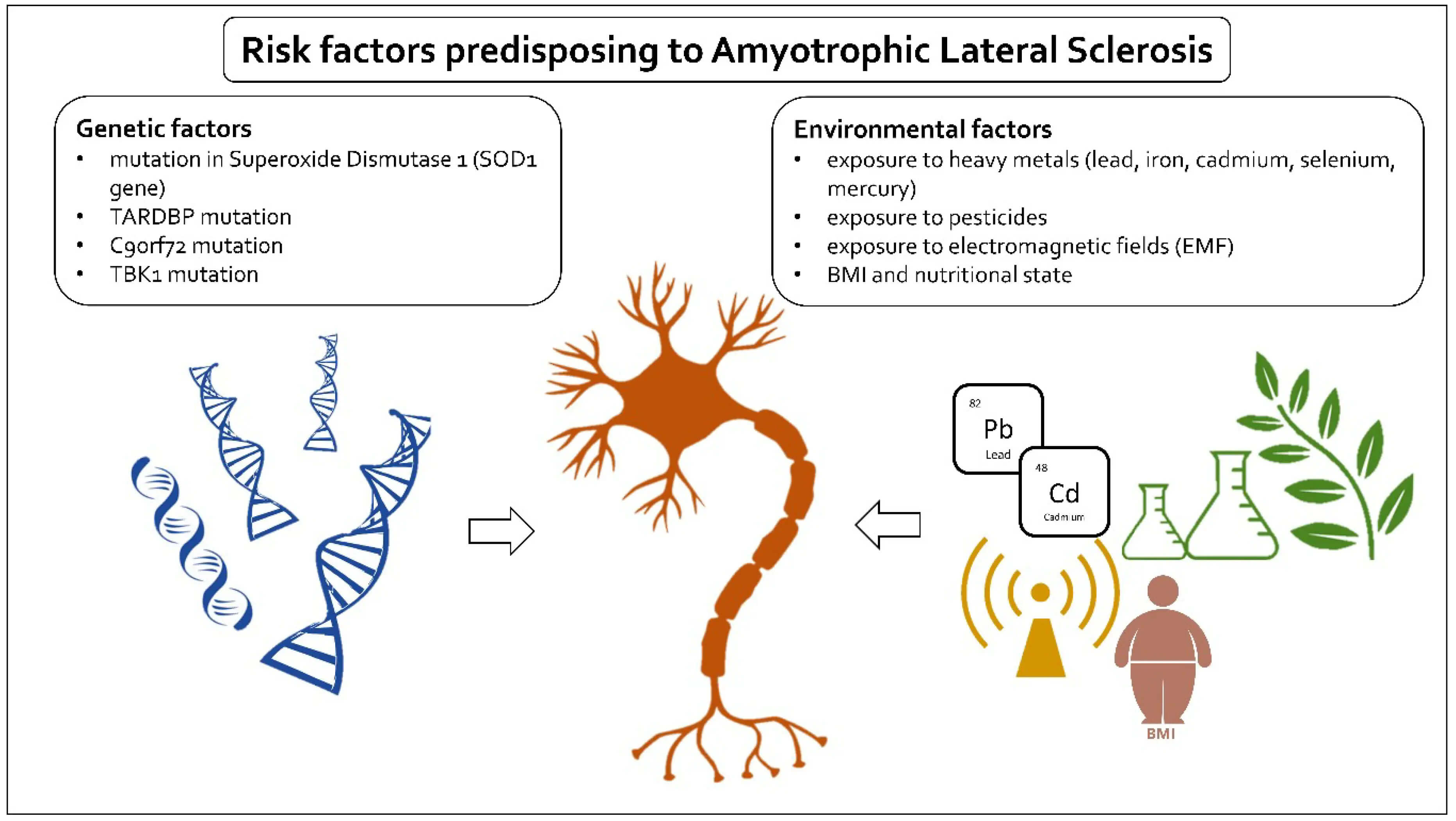
The cause of ALS is not known, and scientists do not yet know why ALS strikes some people and not others. However, scientific evidence suggests that both genetics and environment play a role in motor neuron degeneration and the development of ALS. Research on certain gene mutations suggests that changes in the processing of RNA molecules may lead to ALS-related motor neuron degeneration. RNA molecules are involved with the production of molecules in the cell and with gene activity. Researchers are studying the impact of environmental factors, such as exposure to toxic or infectious agents, viruses, physical trauma, diet, and behavioral and occupational factors.
There is no treatment to reverse damage to motor neurons or cure ALS. However, treatments can help control symptoms, prevent unnecessary complications, and make living with the disease easier. For example, riluzole has been found to modestly prolong survival by about 2–3 months. Gabapentin, pregabalin, and tricyclic antidepressants (e.g., amitriptyline) can be used for neuropathic pain, while nonsteroidal anti-inflammatory drugs, acetaminophen, and opioids can be used for nociceptive pain.
A $1.5 million grant from the U.S. Department of Defense is supporting researchers in the University of Arizona Health Sciences Center for Innovation in Brain Science as they examine whether an investigational drug has the potential to reduce inflammation and increase the regeneration of nerve cells in people with ALS.
Dr. Kathleen Rodgers, associate director of translational neuroscience at the Center for Innovation in Brain Science, is using the two-year grant to study CAP-1902, also known as RASRx1902. Previous research has shown that CAP-1902 reduced inflammation and oxidative stress, improved cognitive function and stimulated muscle regeneration in models of Duchenne muscular dystrophy, another disease that causes progressive muscle degeneration.
RASRx1902 is an investigational oral drug that has been shown to reduce inflammation and oxidative stress (cellular damage that occurs as a consequence of high levels of oxidant molecules), improve cognitive function, and stimulate muscle regeneration in previous studies. Because of its muscle regenerative properties, RASRx1902 has been explored as a potential treatment for Duchenne muscular dystrophy (DMD), a genetic disorder that gradually leads to muscle deterioration. Studies in animal models of DMD have shown the compound increased muscle strength and regeneration, reduced muscle inflammation, degeneration and cell death, without posing any toxic side effects. Based on those promising findings, the U.S. Food and Drug Administration (FDA) granted the designation of orphan drug to RASRx1902 for the treatment of DMD in 2017.
“CAP-1902 has a very promising profile that warrants further development as a treatment for ALS patients,” said Dr. Rodgers, professor of pharmacology in the University of Arizona College of Medicine – Tucson. “This funding from the Department of Defense will enable us to produce CAP-1902 and determine its efficacy as a treatment for ALS, as well as identify blood-based biomarkers of the response to treatment that will help us quickly advance this novel ALS therapeutic to the clinic, where it is needed most.”
The study will target the renin-angiotensin system, a hormone system that regulates blood pressure and inflammatory responses, among other things. The protective arm of the system, which Dr. Rodgers hopes to activate with CAP-1902, is known to reduce inflammation and oxidative stress. The goal is to increase the regeneration of nerve cells and reduce the inflammation that causes nerve cell death and accelerates ALS disease progression.
About orphan drugs
An orphan drug is a drug used to treat, prevent, or diagnose an orphan disease. An orphan disease is a rare disease or condition that affects fewer than 200,000 people in the United States (or less than 5 in 10,000 people in the EU). because they are so rare, they would not be profitable to produce without government assistance. Orphan diseases are often serious or life threatening.
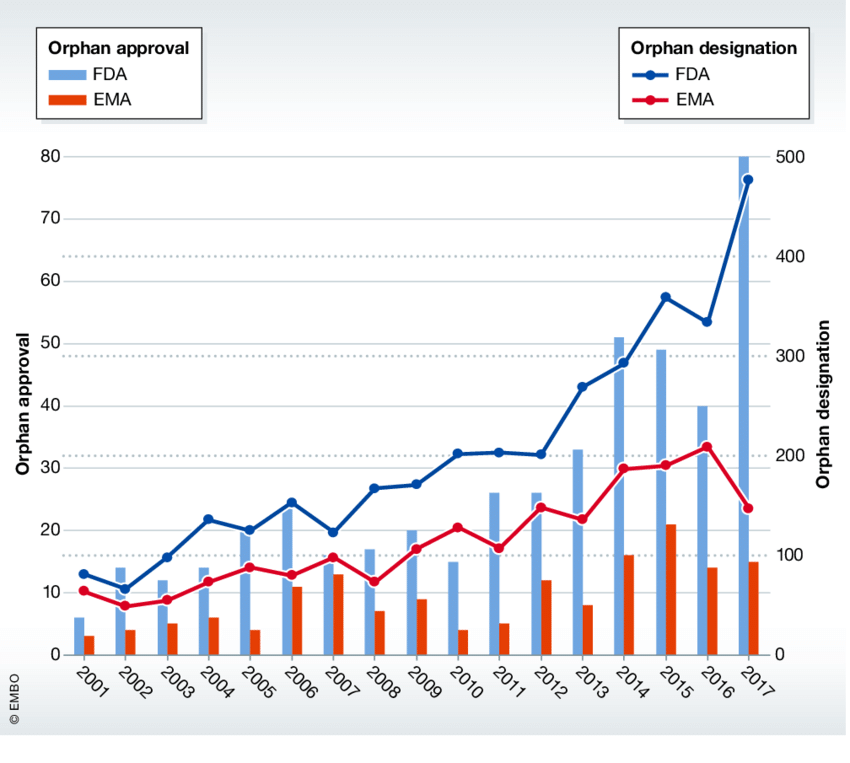
Legislative actions to address this problem, the “Orphan Drug Act” in the United States and the “EU Regulation on Orphan Medicinal Products (Regulation EC 141/2000),” have facilitated the development of orphan drugs by providing both advisory and financial support. At present, there are 2200 drugs designated as orphan drugs that have those benefits assigned to their development, and 160 of them have been authorized for marketing.
Examples of approved orphan drugs
- Revlimid
- Everolimus
- Nilotinib
- Bortezomib
- Pemetrexed
- Dasatinib
- Ivacaftor
- Ruxolitinib
- Sunitinib
- Carfilzomib
- Sorafenib